
News

In A 1st, Doctors In U.S. Use CRISPR Tool To Treat Patient With Genetic Disorder
By: Rob Stein | NPR
Posted on:
Updated at 4:05 p.m. ET
For the first time, doctors in the U.S. have used the powerful gene-editing technique CRISPR to try to treat a patient with a genetic disorder.
“It is just amazing how far things have come,” says Victoria Gray, 34, of Forest, Miss. “It is wonderful,” she told NPR in an exclusive interview after undergoing the landmark treatment for sickle cell disease.
Gray is the first patient ever to be publicly identified as being involved in a study testing the use of CRISPR for a genetic disease.
“I always had hoped that something will come along,” she says from a hospital bed at the Sarah Cannon Research Institute in Nashville, Tenn., where she received an infusion of billions of genetically modified cells. “It’s a good time to get healed.”
But it probably will take months, if not years, of careful monitoring of Gray and other patients before doctors know whether the treatment is safe and how well it might be helping patients.
Sickle cell affects millions of people around the world. About 100,000 are in the U.S., and most of them, like Gray, are African American. A genetic defect causes bone marrow to produce a defective protein that makes blood cells that are sickle-shaped, hard and sticky. The deformed cells get stuck inside blood vessels and don’t carry oxygen normally, causing a host of debilitating and, often, eventually life-shortening complications.
“It’s horrible,” Gray says. “When you can’t walk or lift up a spoon to feed yourself, it gets real hard.”
For the study, doctors are using cells taken from patients’ own bone marrow that have been genetically modified with CRISPR to make them produce a protein that is usually only made by fetuses and by babies for a short time following birth.
The hope is this protein will compensate for the defective protein that causes sickle cell disease and will enable patients to live normally for the rest of their lives.
“It’s exciting to see that we might be on the cusp of a highly effective therapy for patients with sickle cell,” says Dr. David Altshuler, executive vice president, global research and chief scientific officer at Vertex Pharmaceuticals in Boston. Vertex is co-sponsoring the study with CRISPR Therapeutics of Cambridge, Mass.
CRISPR Therapeutics announced the treatment of the first volunteer on Monday but did not name the patient. However, NPR got exclusive access to Gray.
“People with sickle cell disease have been waiting a long time for therapies that just let them live a normal life,” Altshuler says.
“This is a very big deal,” agrees Dr. Haydar Frangoul, medical director of pediatric hematology/oncology at the institute where Gray volunteered. “This could benefit many patients.”
Frangoul’s center, Sarah Cannon, is conducting the study at HCA Healthcare’s TriStar Centennial Medical Center in Nashville, which is one of eight sites recruiting patients for the research in the United States, Canada and Europe. Up to 45 patients ages 18 to 35 will eventually be enrolled.
Other doctors, scientists and bioethicists are also encouraged.
“This is an exciting moment in medicine. CRISPR promises the capacity to alter the human genome and to begin to directly address genetic diseases,” says Laurie Zoloth, a bioethicist at the University of Chicago.
But Zoloth is also cautious. She worries that this and other studies starting up using CRISPR haven’t gone through an extra layer of scrutiny by a panel of outside experts assembled by the National Institutes of Health.

Frangoul acknowledges there are always risks with experimental treatments. But he says the research will go very slowly and carefully with close review by the Food and Drug Administration and other advisory panels.
“We are very cautious about how we do this trial in a very systematic way to monitor the patients carefully for any complications related to the therapy,” he says.
For her part, Gray says she understands there are risks. She also says that she knows the study is a first step and that other patients might only see benefits years from now.
“This gives me hope if it gives me nothing else,” Gray says.
It will probably take several months before doctors detect the first signs of whether the genetically edited cells are producing helpful levels of the protein and even longer to know whether the cells are improving patients’ health. And it will likely take many years to know whether the benefits last a lifetime, as hoped.
Gray, who is married and has four children, was diagnosed with sickle cell disease when she was an infant and started crying during a bath. One major symptom is agonizing, debilitating pain.
Like many sickle cell patients, her symptoms have prevented her from living a full life. She couldn’t play like other children, was afraid to travel and had to give up her dreams of becoming a doctor or a nurse.

The defective blood cells also increase the risk for infections and damage to vital organs such as the heart. They also can cause life-threatening strokes. Many people with sickle cell disease don’t live past their 40s. Gray’s heart has already suffered damage. And that is taking an additional psychological toll.
“It’s scary,” she says, describing how her disease has affected her oldest son, 12-year-old Jamarius.
“He’s older, so he understands. So he started acting out in school. And his teacher told me, ‘I believe Jamarius is acting out because he really believes you’re going to die,’ ” Gray says, choking back tears.
Some patients can get bone marrow transplants, but those procedures are grueling and can be dangerous if the immune system cells produced by the transplanted bone marrow attack their bodies. And most sickle cell patients either don’t have or don’t find a suitable donor.
“It’s real hard,” Gray says. “It was just my religion that just kept me going.”
When she was considering a bone marrow transplant, she heard about the CRISPR trial and jumped at the chance to volunteer.
“I always knew something had to come along and that God had something important in store for me,” Gray says. “It felt like this was meant to be. It was kinda fate. That was an amazing feeling.”
CRISPR enables scientists to make very precise changes in DNA, raising hopes it will lead to new ways to prevent and treat many diseases.
“CRISPR technology has a lot of potential use in the future, not only in blood disorders,” Frangoul says.
Doctors have already started using it to try treat cancer, mostly in China. At least two patients in the U.S. have been treated for cancer, in a study at the University of Pennsylvania in Philadelphia.
Later this year, doctors in Boston are planning to use CRISPR to edit cells in patients’ retinas, in hopes of restoring vision in patients with an inherited form of blindness.
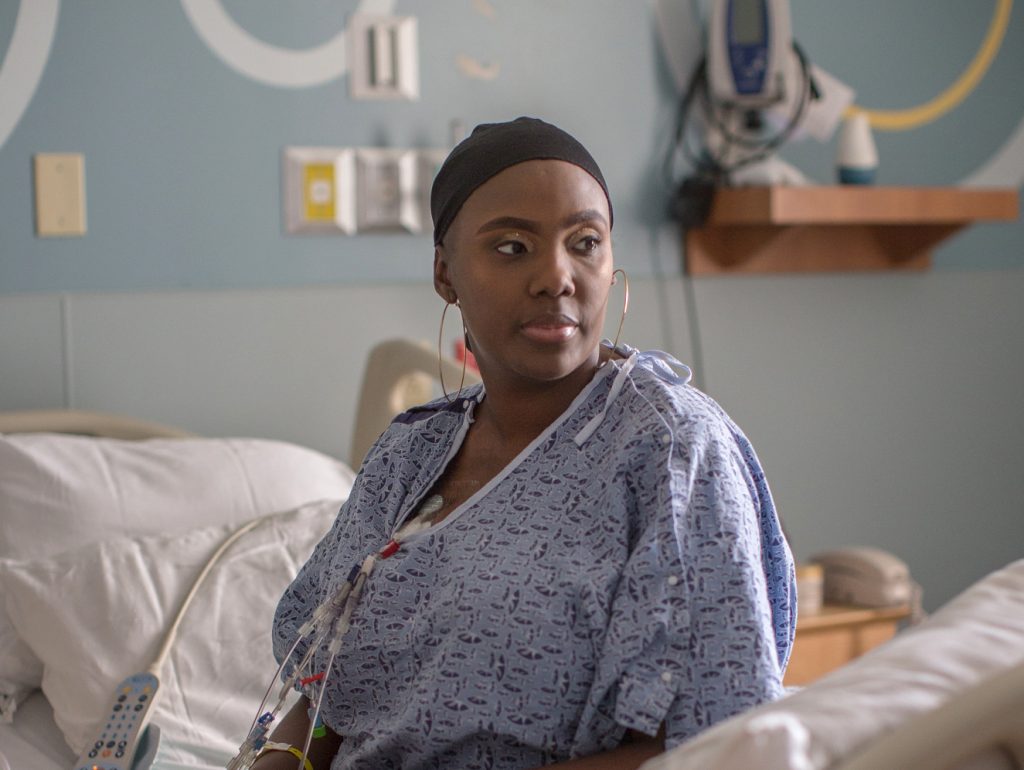
On Monday, CRISPR Therapeutics also revealed the first hint that the approach might be working for beta thalassemia. The beta thalassemia patient’s edited cells have started functioning in the bone marrow, according to the company’s press release.
Moreover, that patient has not required a blood transfusion for more than four months, the company says. Beta thalassemia patients normally require regular transfusions to treat their condition.
Beta thalassemia and sickle cell disease are both caused by genetic defects that cause problems with a protein called hemoglobin. Healthy red blood cells use hemoglobin to carry oxygen throughout the body.
Sickle cell patients’ red blood cells carry defective hemoglobin that deform the cells and don’t transport enough oxygen. The hope is that another form of hemoglobin, known as fetal hemoglobin, will compensate for the defective protein.
Fetal hemoglobin is produced by fetuses in the womb to provide oxygen. In most people, fetal hemoglobin production stops shortly after birth.
“Once a baby is born, a switch will flip on,” Frangoul says. “It’s a gene that tells the red blood cell — the bone marrow cells that produce red cells — to stop making fetal hemoglobin.”
This CRISPR treatment starts with doctors extracting bone marrow cells from patients’ blood. Company scientists then use CRISPR to edit a gene in the cells to make the cells produce fetal hemoglobin.
“That will help the cells make more fetal hemoglobin and make them happier and healthier,” Frangoul says.

The hope is that it will provide a treatment option for all patients, including those who can’t find a matched donor. The approach hopefully will be safer because the cells come from patients’ own bone marrow. So they shouldn’t attack patients’ bodies, Frangoul says.
“This opens the door for many patients to potentially be treated and to have their disease modified to be mild” and to avoid the “horrible, horrible long-term side effects of sickle cell disease,” he says.
For Gray, the process was sometimes long and demanding. It required repeated trips from her home to Nashville for some lengthy stays in the hospital. The chemotherapy was especially rough, leaving her with mouth and throat sores that made it difficult to eat or drink.
“It was really hard and painful for me,” she says.
And Gray was surprised by the discomfort she felt at the moment earlier this month when Frangoul pushed the plunger on a large syringe to infuse more than 2 billion of the edited cells into her body.
“There was a little scary, tough moment for me,” she says. “But after that, I cried. But it was happy tears.”
She was thrilled to finally have what she calls her “supercells” inside her body.
“That’s what all of this is about — the edited cells. It’s the supercells that’s what’s going to make the difference between me having the sickle cell pain crises and not having them,” Gray says.
She hopes it works and imagines a life without sickle cell:
“Being able to wake up and not be in pain. And to just be tired because I’ve done something — and not just tired for no reason. And just to be outside, and jump on the trampoline with my kids.
“And go to graduations and weddings. And see them grow up. That means the world to me.”
9(MDI4ODU1ODA1MDE0ODA3MTMyMDY2MTJiNQ000))